Addressing the Poor Stability Issues in Fusion Protein Cleavage

Addressing Poor Stability Factors During Fusion Protein Cleavage
Measures in preparing your proteins for optimal yield and purity
Introduction
Expression of recombinant proteins as translational fusions is commonly employed to enhance stability, increase solubility, and facilitate purification of the desired protein. Affinity tags are appended to proteins so that they can be purified from their crude biological source using an affinity technique. Affinity resins are available as capture reagents to purify these tagged proteins. Affinity tags that are commonly fused to proteins of interest using various expression vector systems, include (polyhistidine), FLAG (DYKDDDDK), glutathione S-transferase (GST), and Myc tags (1).
In general, such fusion proteins must be cleaved to separate the fused tag and release the mature protein in its native form (2). Purification and on-column fusion protein cleavage methods have been developed to isolate recombinant, untagged proteins in a single chromatographic step. Use of a GST-fusion partner in conjunction with glutathione affinity media and application of an on-column cleavage strategy is one of the most effective methods in terms of simplicity, flexibility, robustness, efficiency, and fusion protein purity (3,4,6).
However, all fusion protein systems present an inherent problem - the fusion partner (affinity tag) is often difficult to remove. Most systems rely on proteolytic cleavage to separate the fusion partner from the target protein. Several problems may be encountered during proteolytic cleavage, including:
- spurious, non-specific proteolytic attack of the fusion protein;
- the need for elevated temperatures for efficient cleavage, which can denature or cause aggregation of the fusion protein;
- incomplete cleavage, which reduces the yield and/or introduces heterogeneity in the purified protein;
- the need for additional steps to separate the cleaved fusion protein from the fusion tag, deactivate and remove the protease, and exchange buffer or desalt.
The pGEX E. coli expression vectors, available as part of the GST Gene Fusion system, encode for N-terminal GST molecules followed by protease cleavage sites in all three reading frames and with three different protease cleavage sites. In this study, pGEX-6P-1, pGEX-5X-1, and pGEX-2T which encode optimal recognition sites for PreScission Protease, factor Xa, and thrombin, respectively, have been used.
The performance of PreScission Protease, factor Xa, and thrombin was compared by screening their enzymatic stability, efficiency in removing the GST-fusion partner, and proteolytic specificity on cleavage of a diverse range of fusion proteins.
Preparation of GST-fusion proteins and binding to GSTrapTM FF column
PreScission Protease, recombinant polyhistidine-tagged factor Xa, and thrombin, (2 units enzyme/mg of bound fusion GST-fusion protein) were diluted in the appropriate cleavage buffer equal to 90% of the volume of the two 5 ml GSTrapTM FF columns. The enzymes were injected into the columns at a flow rate of ~5–7 ml/min using a syringe. Following injection, the column was placed in a closed flow status and the system was incubated online for 3–18 h at 4–22°C according to the proteolytic enzyme used.
The Spin-Column Based Method
The easiest and safest method, readily available in a kit format, is the spin-column based method. The binding element in spin-column systems is usually composed of glass particles or powder, silica matrices, diatomaceous earth, and ion exchange carriers. In this method, nucleic acid binding is optimized with specific buffer solutions and extremely precise pH and salt concentrations. Sample lysates are passed through the silica membrane using centrifugal force, with the RNA binding to the silica gel at the appropriate pH. The membrane containing residual proteins and salt is then washed to remove impurities, and flow-through is discarded. RNA is subsequently eluted with RNase-free water.
Elution of cleaved fusion protein
Prior to elution, an auxiliary 1 ml HiTrap column was connected downstream of the 5 ml GSTrapTM FF proteolytic cleavage columns, in-line with the fraction collector. The auxiliary column was matched to the protease used for cleavage. The column used was either GSTrapTM FF, Ni2+-charged HiTrap Chelating HP, or HiTrap Benzamidine FF (high sub) with specific affinity for PreScission Protease (GST-fused enzyme), recombinant polyhistidine tagged factor Xa, or thrombin, respectively. Each column was pre-equilibrated with the appropriate cleavage buffer specific for the enzyme used.
The auxiliary column is beneficial for:
- Detaining any cleaved material upon flow start-up, which minimizes loss of cleaved product and allows for rapid baseline recalibration before peak elution.
- Filtering and removing the proteolytic enzyme from the released target protein.
Elution of the released target protein occurred immediately upon flow start-up (flow rate of ~1 ml/min). Following elution and the return of absorbance to the baseline, the GST-affinity peak was eluted with elution buffer (optimized buffer for each enzyme) containing 10 mM reduced glutathione applied in a one-step gradient (100% elution buffer).
The GST moiety, any uncleaved GST-fusion protein, endogenous E. coli proteins that have affinity for glutathione, and PreScission Protease, if it was used for cleavage, are all eluted in this step.
Eluted protein fractions were analyzed with SDS-PAGE according to standard procedures (7) and protein quantitation was determined using CoomassieTM Protein Assay Reagent (Pierce).
Temperature-dependence of proteolytic cleavage
The effect of temperature on cleavage of GST-p52 fusion protein was tested using a 12 h digestion period (Fig. 1). Each enzyme was tested at 4°C, 12°C, and 22°C, the optimum temperatures for PreScission Protease, factor Xa, and thrombin, respectively.
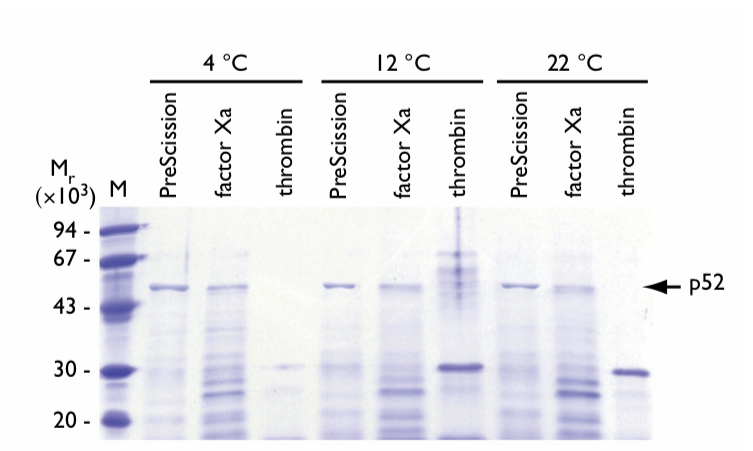
Fig.1. SDS gel electrophoresis of eluates following on-column cleavage of GST-p52 fusion protein at different temperatures. The incubation time was 12 h. Proteases and incubation temperatures are shown above the lanes. The arrow indicates the location of the released p52 protein. M = molecular weight markers.
PreScission Protease shows stable proteolytic processing at 4°C with the highest enriched fraction migrating to the appropriate theoretical molecular weight of the cleaved p52 fusion protein. At 12°C and 22°C, an increase in degraded p52 protein was observed relative to cleavage reactions at 4°C.
Factor Xa cleavage significantly increases degradation of the p52 protein with increasing temperature. When the temperature was elevated to 12°C and 22°C, the band corresponding to p52 protein decreased in intensity and shifted to a lower molecular weight breakdown product of ~Mr 26,000.
Thrombin cleavage is temperature dependent with poor cleavage occurring at 4°C. The majority of cleaved p52 protein was in the form of a breakdown product migrating at ~Mr 32,000. In addition, aggregation and precipitation of p52 occurred upon elution from the column. At elevated incubation temperatures for thrombin, cleavage activity was increased. Aggregation and proteolytic breakdown of p52 protein occurred and the formation of a stable p52 degradation product with ~Mr 32,000 was observed. Upon sample loading on the SDS polyacrylamide gel, cleaved p52 protein aggregated and precipitated, necessitating resolubilization prior to electrophoresis.
Time-dependent proteolytic cleavage
The effect of incubation time on cleavage of GST-p40 was examined using each protease at its optimum temperature (Fig. 2). Samples were incubated for 6, 12 and 18 h.
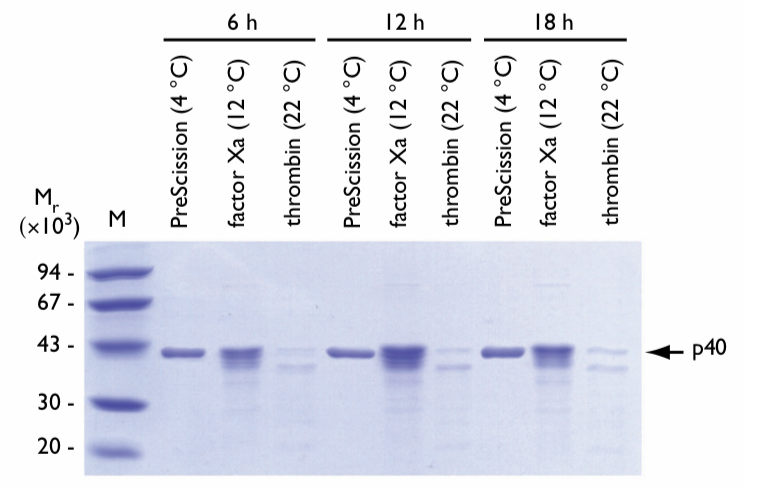
Fig. 2. SDS gel electrophoresis of eluates following on-column cleavage of GST-p40 fusion protein for different incubation times at optimum temperatures. Proteases, optimum incubation temperatures, and incubation times are shown above the lanes. The arrow indicates the location of the released p40 protein. M = molecular weight markers.
Longer incubation times did not affect results when using PreScission Protease at its optimum temperature.
Factor Xa displayed a slight increase in p40 protein degradation with longer incubations. More p40 breakdown products were observed after each successive incubation period.
Thrombin cleavage resulted in immediate spurious proteolytic attack on the p40 protein. The primary breakdown product yields (~Mr 37,000) appeared after 6 h of incubation. After 18 h, the yield of the breakdown product is approximately equal to that of intact p40 protein. In addition, observed levels of p40 protein were significantly decreased following thrombin cleavage because of protein aggregation and precipitation upon elution from the column.
pH-dependent proteolytic cleavage
The effect of pH on cleavage of GST-p28 was tested at three pH values using a 12 h digestion (Fig. 3). Each enzyme was tested at its optimum temperature at pH 7.4 (phosphate buffered saline), 7.7 (HEPES), and 8.0 (Tris-HCl).
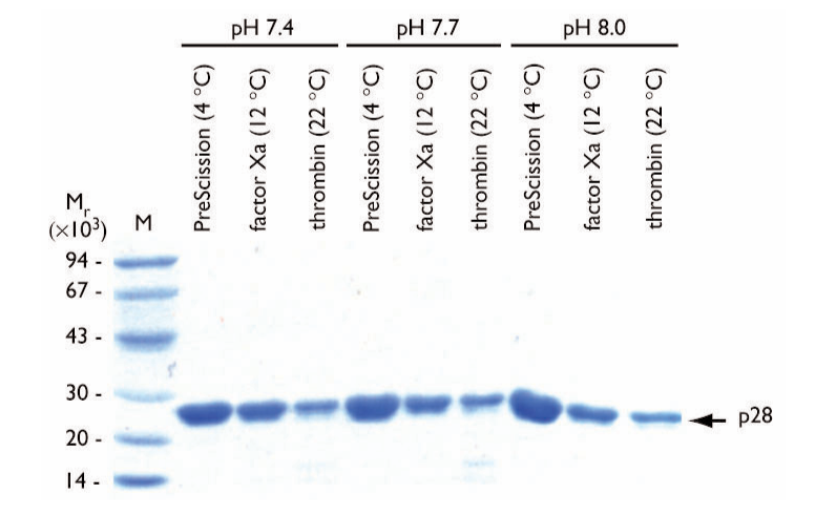
Fig. 3. SDS gel electrophoresis of eluates following on-column cleavage of GST-p28 fusion protein at different pH values at optimum temperatures. The incubation time was 12 h. Proteases and pH values are shown above the lanes. The buffers used at each pH value were phosphate buffered saline, pH 7.4; HEPES, pH 7.7, and Tris-HCl, pH 8.0. The arrow indicates the location of the released p28 protein. M = molecular weight markers.
pH did not affect purity or concentration of the final p28 protein during cleavage with PreScission Protease.
For factor Xa or thrombin, pH did not considerably affect the final p28 protein in terms of homogeneity. The final yield of p28 is, however, generally reduced compared with normalized protein levels due to aggregation and precipitation of the p28 protein after on-column cleavage.
Salt-dependent proteolytic cleavage
Salt sensitivity of the cleavage reactions was tested at two commonly used salt concentrations; 30 mM (NH4)2SO4 and 300 mM NaCl. Digestions were performed at optimum temperatures for the proteases for 6 h with GST-p71 as substrate (Fig. 4).
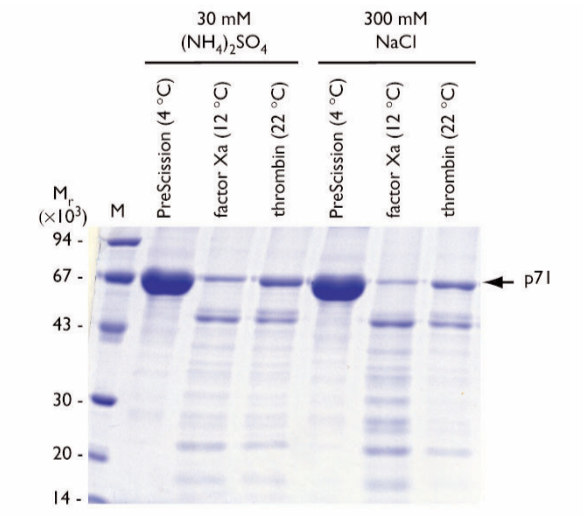
Fig. 4. SDS gel electrophoresis of eluates following on-column cleavage of GST-p71 fusion protein at different salt concentrations at optimum temperatures. The incubation time was 6 h. Proteases and salt concentrations are shown above the lanes. The arrow indicates the location of the released p71 protein. M = molecular weight markers.
Under these conditions, PreScission Protease did not affect the homogeneity of the final p71 protein product in the presence of the selected salts.
Factor Xa cleavage reduced the level of soluble cleaved p71 protein relative to PreScission Protease, due to cleaved p71 protein aggregation and precipitation following column elution. A stable p71 break- down product of ~Mr 53,000 was observed.
Thrombin cleavage also reduced the level of soluble cleaved p71 protein relative to PreScission Protease. The presence of 30 mM (NH4)2SO4 appears to be more effective in terms of soluble p71 protein yield. As in the case of the factor Xa-cleaved product, a stable p71 breakdown product of ~Mr 53,000 was observed.
Additive-dependent proteolytic cleavage
The on-column cleavage strategy was used to examine the effect of additives on cleavage of GST-p105 fusion protein (Fig. 5). The additives used were 2 mM DTT, 2% (v/v) glycerol, or 0.1% (v/v) TritonTM X-100. Cleavage reactions were incubated at the optimum time and temperature for each enzyme (12 h, 4°C for PreScission Protease; 6 h, 12°C for factor Xa; 3 h, 22°C for thrombin).
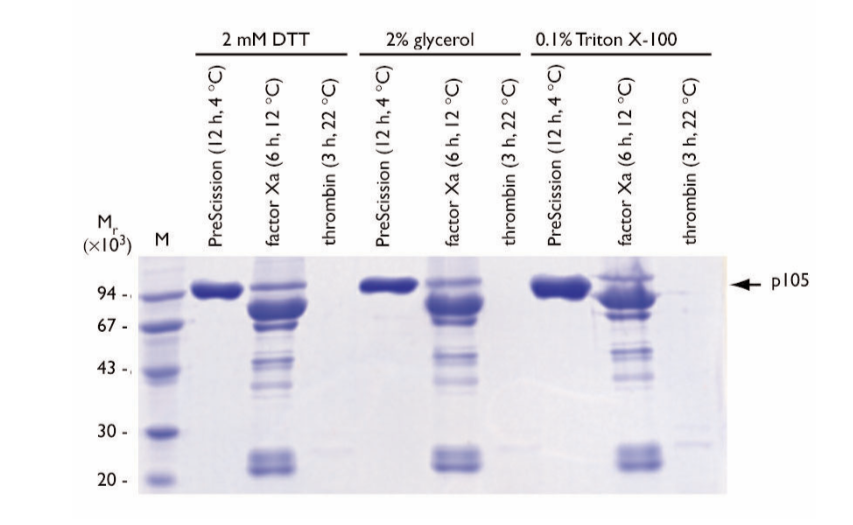
Fig. 5. SDS gel electrophoresis of eluates following on-column cleavage of GST-p105 fusion protein in the presence of various additives. Cleavage reactions were incubated at the optimum time and temperature for each enzyme as indicated above each lane. The arrow indicates the location of the released p105 protein. M = molecular weight markers.
Negligible differences among the additives were observed in the final p105 protein product following cleavage with PreScission Protease. In the presence of 0.1% (v/v) Triton X-100, the PreScission Protease cleaved p105 protein displayed a diffuse migration pattern on the gel.
Factor Xa cleavage in the presence of the additives resulted in low yields of p105 protein. A common degradation pattern was observed, with a predominant degradation product of Mr ~75,000.
Thrombin cleavage reactions displayed poor solubility following on-column cleavage and elution. Soluble, cleaved p105 protein was not detected following thrombin cleavage in the presence of the additives. In the presence of 0.1% Triton-X-100, a minor soluble p105 protein breakdown product migrating at Mr ~75,000 was observed. Additional breakdown products migrating at Mr ~30,000 were also detected. The Mr ~75,000 degradation product is similar in size to the product observed in the Factor Xa experiments. The remaining cleaved p105 protein material was precipitated on-column or immediately following elution in all experiments with thrombin.
Conclusion
Recombinant proteins are commonly expressed as translational fusions to Glutathione S-transferase (GST) for facilitating large-scale expression and purification. Removal of the GST tag is often necessary to be able to perform functional or structural studies of the target protein. In this study three protease sites (PreScission protease; factor Xa; and thrombin) were tested on a variety of target proteins to elucidate a trend in proteolytic behavior.
Although protease digests have to be optimized for each protein of interest, some general conclusions can be made.
Performing the digest at optimum temperature is crucial for efficient cleavage. PreScission protease was superior to the other enzymes tested, primarily for its activity at 4°C, making this protease a good choice when working with temperature-sensitive proteins.
Thrombin performed the poorest in these sets of experiments, demonstrating that non-specific cleavage is more likely to take place with this enzyme. Thrombin also possesses a relatively high temperature optimum (22°C), which can adversely affect protein stability.
With factor Xa, problems concerning specific cleavage were frequently observed.
Salt concentrations and pH values tested in this study did not make a significant difference in the proteolytic behavior of these proteases, indicating a relatively high tolerance for variation in these parameters.
The cleavage conditions are more dependent on the protein of interest and should be optimized for each target protein separately taking into consideration protein instability, cleavage efficiency, aggregation, and purity of the final cleaved protein product.
-It is your turn now-

References
- Kimple ME, Brill AL, Pasker RL. Overview of affinity tags for protein purification. Curr Protoc Protein Sci. 2013;73:9.9.1-9.9.23. Published 2013 Sep 24.
- Coligan,J.E., Dunn,B.M., Ploegh,H.L., Speicher,D.W. and Wingfield,P.T. (eds) (2001) Current Protocols in Protein Science, Vol. 2. Wiley, New York.
- Cordingley M. G. et al., J. Biol. Chem. 265, 9062–9065 (1990).
- Walker P. A. et al., BioTechnology 12, 601–605 (1994).
- Ausubel, F. M. et al. (eds.), Short Protocols in Molecular Biology, John Wiley & Sons, New York, (1992).
- Knaust, R. et al., Life Science News 6, 12–13 (2000).
- Fling, S. P, and Gregerson, D. S., Anal. Biochem. 155, 83–88 (1986).