The Rise of Cryo-EM Among Structural Characterization Methods: NMR, X-Ray Crystallography

The Rise of Cryo-EM Among Structural Characterization Methods
Reviewing NMR, X-Ray crystallography and electron microscopy
Introduction
Structural biology is the study of the molecular structure and dynamics of biological macromolecules, by utilizing techniques and principles of molecular biology, biochemistry and biophysics. In order to determine the spatial relationship of the hundreds of thousands of atoms, and to follow the changes in their relative locations within a biological macromolecule, multiple methodologies with very different physical principles, such as X-ray crystallography, NMR spectroscopy, cryo-electron microscopy (cryo-EM), X-ray solution scattering, neutron diffraction, and other spectroscopic techniques have been implemented. Amongst these, X-ray crystallography, NMR, and (cryo-EM) represent the three main methods widely employed to reveal structural information pertaining to a large variety of macromolecules.
X-Ray Crystallography
Principle:
Given its ability to resolve structures of macromolecules at atomic resolution, X-ray crystallography is the most powerful tool in modern structural biology. This technique uses X-rays to determine the position and arrangement of atoms in a crystal. Its foundation principle lies in Bragg’s law of X-ray diffraction by crystals, i.e. by well-ordered packing of homogenous molecules in three-dimension. Illuminated by a beam of X-ray light, the crystal can diffract the light at various angles, some of which have stronger intensity than others (Fig. 1). This kind of intensity variation at different angles can be recorded on media as a “diffraction pattern”. The diffraction pattern, normally appearing as a series of sharp spots, reflects the structural arrangement of atoms within the crystal and therefore can be used to deduce the original structure of the crystal using Bragg’s Law.
In order to solve the structure of the molecule of interest, besides the measured intensities of the diffraction spots, additional information called phases of the spots is required which is obtained by other experimental or computational means. The intensity and phase information of multiple diffraction patterns of the crystal can then be reconstructed and Fourier transformed in a computer to generate a virtual structure for interpretation.
The quality of the structure heavily depends on the sharpness of the diffraction spots, which in turn is determined by the degree of order of the crystal.
Therefore, modern X-ray crystallography’s essential step is to obtain highly-ordered three-dimensional crystals. In order to achieve this, a large amount of highly purified macromolecules may be necessary when screening a large number of crystallization conditions. Also engineering of the molecules, e.g. stability promotion, side-chain modification, proteolysis, may be important to improve the crystal quality.
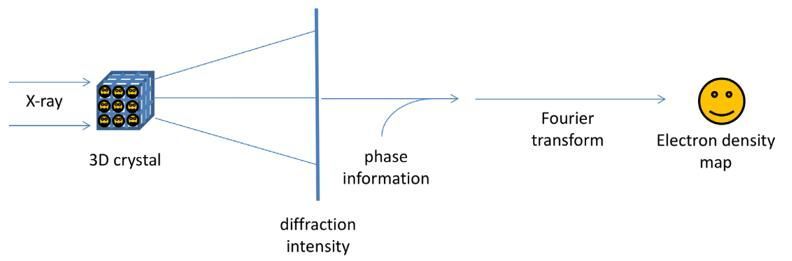
Fig.1. The principle of X-ray crystallography
Workflow:
The technique of single crystal X-ray crystallography has three basic steps. The first and usually most difficult step is to produce an adequate crystal of the studied material. The crystal should be sufficiently large with all dimensions larger than 0.1 mm, pure in composition and regular in structure, and have no significant internal imperfections such as cracks or twinning. The crystal is subsequently placed in an intense beam of X-rays, usually of a single wavelength, to produce a regular reflection pattern. The angles and intensities of diffracted X-rays are measured with each compound having a unique diffraction pattern. Previous reflections disappear and new ones appear along with the gradual rotation of the crystal, and the intensity of every spot is recorded at every orientation of the crystal. Multiple data sets may have to be collected since each set covers slightly more than half a full rotation of the crystal and typically contains tens of thousands of reflections. Ultimately, these collected data are combined computationally with complementary chemical information to obtain and refine a model from the arrangement of atoms within the crystal (Fig. 2). The final refined model of the atomic arrangement is called a crystal structure and is usually stored in a public database.
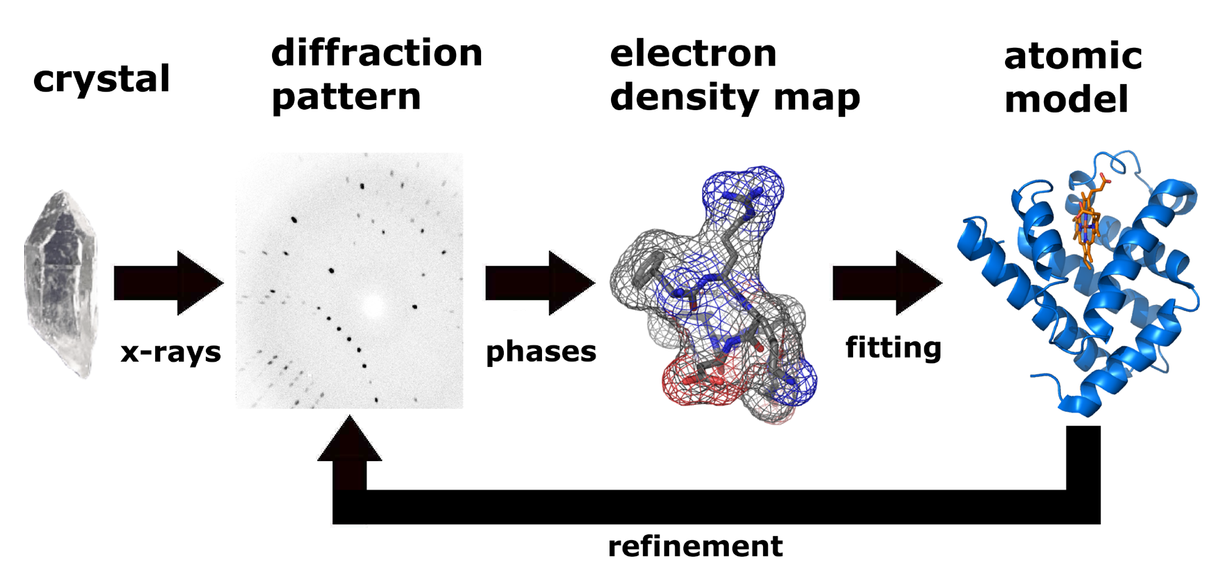
Fig. 2. Workflow for solving the molecular structure using X-ray crystallography.
Advantages:
- Most advanced method available for obtaining high-resolution structural information.
- High atomic resolution.
- Not limited by molecular weight of the sample.
- Suitable for water-soluble proteins, membrane proteins and macromolecular complexes.
- Can reveal considerable information about the mobility (dynamics) and heterogeneity of protein structures.
Disadvantages:
- Sample must be crystallizable.
- Crystallization of biological macromolecules with high molecular weight can be difficult. Particularly, membrane proteins are more challenging to crystallize because of their large size and relatively poor solubility.
- An organized single crystal must be obtained to allow appropriate diffraction.
- Obtained three-dimensional structure of biological sample only represents a static form of the tested molecule (one of many possibilities), rather than a dynamic one.
NMR Spectroscopy
Principle:
The nuclei in the atoms in materials are spinning and charged and, as such, most form magnetic dipoles (hence the nuclear magnetic in NMR). As a simple explanation, these dipoles are often compared with little bar magnets with north and south poles (Figure 3A). Normally these nuclear bar magnets are randomly oriented. The result is that no net magnetic field arises from them and thus no NMR signal can be generated. However, when placed in a strong magnetic field, Bo , on average some of the nuclei “align” with Bo . The result is a net magnetic moment, M, from the sample (Figure 3B).
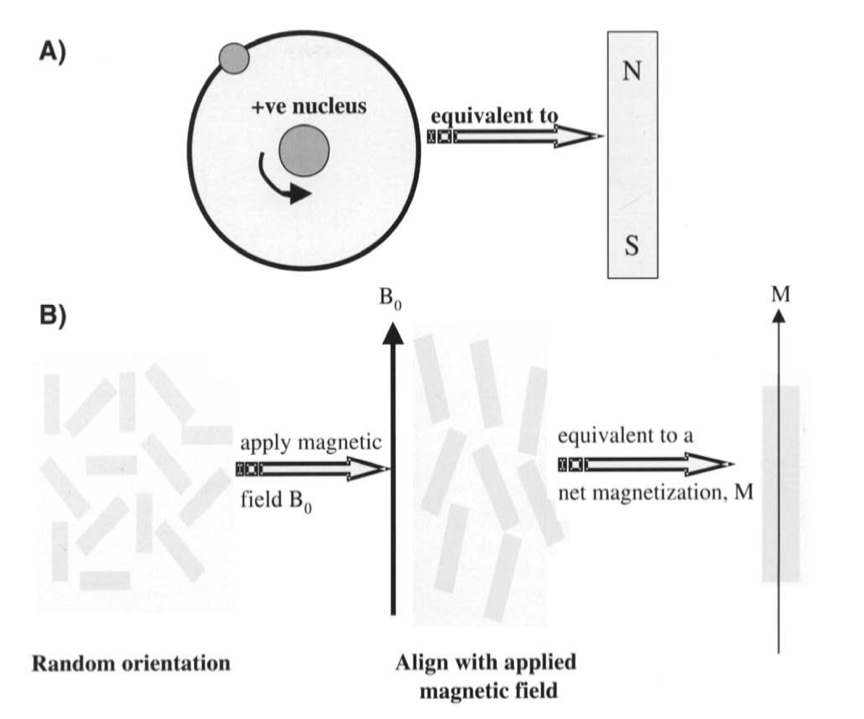
Fig 3. (A) Nuclear dipole equated to a bar magnet. (B) When placed in a magnetic field, Bo, the nuclear moments align with Bo and generate a net magnetic moment, M.
Nuclei in differing chemical environments (different chemical structures) bond to other nuclei by the sharing of electrons; thus, different chemical groups are subsequently surrounded by different electron shells. These electrons also have associated magnetic fields that have the effect of shielding the nuclei from the applied Bo by slightly differing amounts, depending on the number of electrons in the shell. Consequently, nuclei in different chemical groups will resonate at slightly different frequencies, an effect referred to as the chemical shift. When excited, complex chemical samples thus generate multiple signals at different frequencies corresponding to all of the distinct chemical groups (Fig. 4.) that are detected simultaneously in the signal. A technique called the Fourier transform is used to work out the frequencies of the signal, generating a NMR spectrum. This is the basis of NMR spectroscopy, and the information contained within the NMR spectrum can be used to determine the chemical makeup and structure of materials.
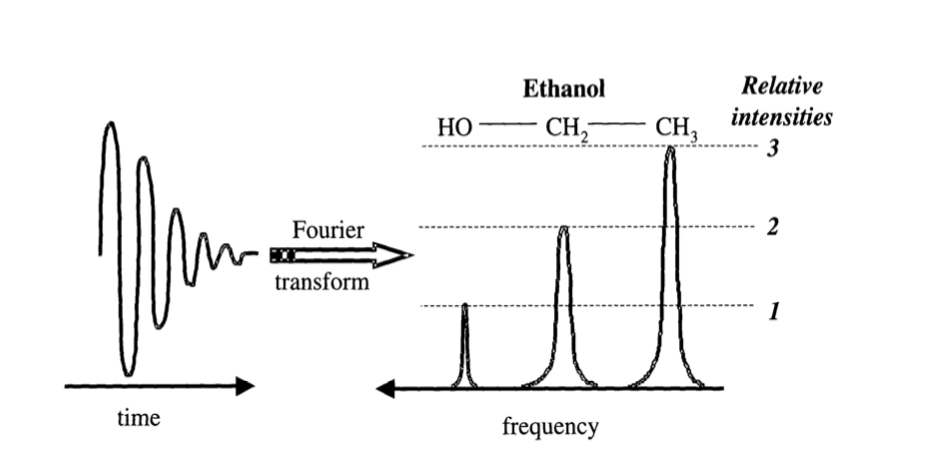
Fig. 4. Distinct chemical groups emit signals at different frequencies.
Workflow:
There are four main steps in an NMR experiment: sample preparation, data acquisition, spectral processing, and structural analysis. NMR analysis is performed on aqueous samples of protein with high purity, high stability, and high concentration. A sample volume ranging from 300 to 600 μL with a concentration range of 0.1-3 mM. The use of stable isotopes 15N, 13C and 2H for protein labeling can effectively increase signal intensity and resolution. Selective labeling of certain amino acids or chemical groups of proteins can greatly reduce signal overlap. Multidimensional NMR experiments are utilized to acquire information about the protein (Fig. 5). The spectral processing is then performed to determine the atoms of the protein corresponding to each spectral peak on different NMR spectra. Finally, a series of spatially structured information such as NOE and J coupling constants are used to calculate the spatial structure using distance geometric or molecular dynamics methods.
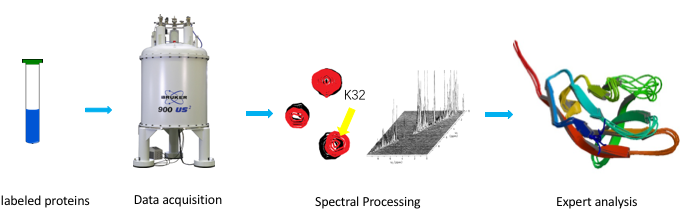
Fig. 5. Workflow of NMR spectroscopy
Although the amount of three-dimensional structure data of proteins obtained by NMR technology is not comparable to that of X-ray crystallography, the unique advantages of NMR technology have been widely noticed: NMR is able to provide information on a kinetic basis, such that the internal movement of proteins over multiple time scales and their binding mechanism to ligands can therefore be solved. Currently, with continuing advancements in magnet and gradient technology coupled with increasingly powerful computers, NMR spectroscopy and imaging can be used to investigate a wide range of biological processes in systems as diverse as a single cell, isolated perfused organs, and tissues in vivo.
Advantages:
- Dynamic technique.
- Flexibility - wide range of biological processes can be investigated.
- Non-destructive and non-invasive.
- Three-dimensional structures in their natural state can be measured directly in solution.
- Can provide unique insights into dynamics and intramolecular interactions.
- Macromolecular three-dimensional structure resolution can be as low as sub-nanometer.
Disadvantages:
- Limited to small (<40 kDa), stable, soluble proteins that do not aggregate at the high concentrations required for data collection.
- Large amounts of pure samples are needed to achieve an acceptable signal to noise level.
- Highly sensitive to motion which can lead to signal distortions and artifacts.
- The high-magnetic field can cause problems with other equipment in a laboratory. Therefore, extra precautions may need to be taken, especially if working space is limited.
Cryo-EM:
Principle:
In this method, fully preserved samples are imaged by freezing them in a thin layer of a non-crystalline form of solid water, called amorphous or vitreous ice. Given that vitreous ice is maintained at liquid nitrogen temperatures, this technique was termed ‘cryo-EM’. Although we mainly associate the term cryo-EM with macromolecular electron microscopy, the 3D study of macromolecules, viruses, organelles and cells using a transmission electron microscope (TEM), cryo-EM is a general term for the observation of low-temperature specimens using an electron microscope. This term can also be used for the observation of frozen material by a scanning electron microscope (SEM), which produces vastly different images from those of a TEM.
Electron microscopes are imaging devices that use electrons instead of light and electromagnetic lenses instead of glass lenses to produce magnified images of an object being studied. Electrons can either pass through the sample, and in doing so get slightly scattered (as in TEM), or ‘bounce off ’ the sample (as in some forms of SEM). The interaction of these electrons with the sample gives rise to various types of images. Macromolecules imaged by a TEM in ice are referred to as projections and can be thought of as a sum of the density through the macromolecule from a particular orientation, similar to looking through one’s hand in an X-ray picture taken at a hospital. Due to the scattering nature of electrons, a very high vacuum is required inside the microscope which means that biological specimens have to be stabilized accordingly.
Workflow:
A typical cryo-EM workflow includes sample preparation, low dose data acquisition, and model building. Before using Cryo-EM to observe the sample, negative staining EM can be utilized for rapid screening of homogeneous sample. The Cryo-EM single particle analysis technique begins with sample vitrification. During this process, the protein solution is instantly cooled, so that the water molecules do not crystallize, forming an amorphous solid. The frozen sample is then screened and data is collected in the system. A series of two-dimensional images can be taken during this period. Next, based on plenty of two-dimensional images acquired, particle alignment and classification are carried out. In the end, the data is processed by reconstruction software to generate a three-dimensional structural model (Fig. 6).
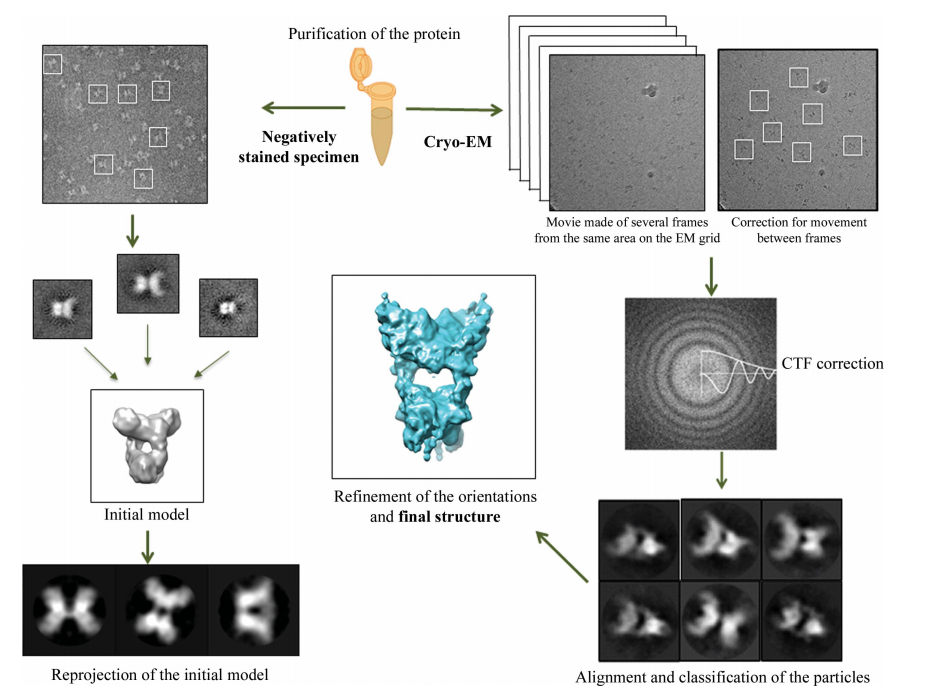
Fig. 6. Schematic representation of three-dimensional single-particle reconstruction using Cryo-EM.
Advantages:
- Rapid freeze treatment of the sample maintains it closer-to-native state.
- Small amount of sample (0.1 mg) required.
- Protein does not need to crystallize.
- Poses fewer restrictions on sample purity.
Disadvantages:
- The low signal-to-noise ratio (or low contrast) of the captured images limits the size of the macromolecules observed (ideally these should be larger than 250 kDa).
- Electron beam-induced movement during exposure results in degradation of the image quality.
Summary of the Advantages and Limitations of Each Structural Biology Technique
X-ray crystallography
| Advantages | Limitations | Target molecules | Resolution |
|---|---|---|---|
| Well developed | Difficult for crystallization | Soluble proteins | High |
| High resolution | Difficult for diffraction | membrane proteins | |
| Broad molecular weight range | Solid structure preferred | ribosomes | |
| Easy for model building | Static crystalline state structure | DNA/RNA | |
| protein complexes |
NMR
| Advantages | Limitations | Target molecules | Resolution |
|---|---|---|---|
| High resolution | Need for high sample purity | MWs below 40–50 kDa | High |
| 3D structure in solution | Difficult for sample preparation | Water soluble samples | |
| Good for dynamic study | Difficult for computational simulation |
Cryo-EM
| Advantages | Limitations | Target molecules | Resolution |
|---|---|---|---|
| Easy sample preparation | Relatively low resolution | >250 kDa | Fast approaching high resolution (typically sub 4 Å) |
| Structure in native state | Applicable to samples of high molecular weights only | Virions, membrane proteins, large proteins, ribosomes, complex compounds | |
| Small sample size | Highly dependent on EM techniques | ||
| Costly EM equipment |
The Rise of Cryo-EM
For many years, structure determination of biological macromolecules by cryo-EM was limited to large complexes or low-resolution models. With recent advances in electron detection and image processing, beginning in early 2013, the resolution of cryo-EM is now starting to rival that of X-ray crystallography. These advances are provided by two major innovations. One is the employment of a direct electron detector (DED) for electron microscopy. DED can detect electrons directly and read them at high frame rate without a mechanical shutter. Their higher performance is due to greatly improved quantum efficiency as compared to previous generations of detectors. Motion correction has become the standard to compensate for the blurring effect of stage drift and beam-induced movement. The other is advancement of image processing methods and the constant increase of microprocessor performance, which allow accurate classification of hundreds of thousands of EM images with computationally expensive algorithms.
These two technologies have led to a “resolution revolution” with atomic structures no longer being the exclusive prerogative of X-ray crystallography or NMR spectroscopy, and have made cryo-EM an important tool to analyze the structure of dynamic biomolecules.
The three-dimensional structures of biological molecules provide great insights into the laws of life activities and mechanisms of diseases, and thereby allow rational design of novel diagnostic and therapeutic agents. Cryo-EM is gradually adding to a better structural understanding of various complex systems and even helping resolve structures that have proven intractable to other methods. One such area is the working of the respiratory chain. Answering questions as to how the proton gradient is harnessed by ATP synthase to produce ATP, and resolving structures of large protein complexes such as cytochrome bc1 and complex I, as well as super-complexes such as the 1.7 MDa respirasome have been possible due to cryo-EM analysis. Our understanding of several important neurodegenerative diseases has rapidly changed recently with a series of landmark papers detailing the structure of tau and amyloid-b proteins facilitated by cryo-EM analysis. The structures reveal some unprecedented details in this important class of proteins.
Although still limited in resolution compared to both X-ray crystallography and NMR, cryo-EM can provide valuable insights into inhibitor binding and therefore open up new avenues for structure-based drug design pipelines. For instance, since 2013, ~50 new single-particle cryo-EM structures have been determined from all the major clades within the transient receptor potential channel membrane-protein family where there had been a paucity of structural information on the full channels. Another example is of the yeast isoform of imidazoleglycerolphosphate-dehydratase, an essential enzyme in histidine biosynthesis, which is more sensitive to small-molecule inhibitors and intractable to crystallization, has recently been determined by cryo-EM. The power of cryo-EM when tackling proteins from a native source has been highlighted by the recently solved structure of the malarial translocon to 3.5 Å, which can now effectively support inhibitor design. N
The large amount of screening space that is required to find a suitable crystallization condition can be a limiting factor when using X-ray crystallography for structure determination. For large protein complexes this becomes more challenging, and specialized cellular machinery may be required for the synthesis of sufficient protein for extraction from host tissue. In contrast, cryo-EM is less demanding on the quantity of sample than a typical X-ray or NMR experiment. An important challenge with cryo-EM are the high costs of the purchase and maintenance of the powerful microscopes that are required to elucidate high-resolution structures. The computation-time requirements of the image-processing pipeline are also important and require supercomputers. Poor access to these instruments (in relation to the growing demand) and their costs call for coordinated funding models and group use. Also, correct sample preparation, image collection and analysis is not as easy as it sounds and appropriate training is required.
Moreover, even with recent developments in cryo-EM, the poor signal to noise in the raw images of sub-65 kDa molecular masses makes particle identification and alignment a significant challenge. On the other hand, in X-ray crystallography, multi-protein complexes, particularly those which involve membrane proteins or where the complexes are not stable over the crystallization time scale, present an important limitation.
Atomic resolution is critical to defining the minute structural details that can clearly elucidate enzyme mechanisms and thereby improve disease understanding and support inhibitor design for drug discovery. Therefore, the use of cryo-EM as a complementary tool to X-ray crystallography to obtain structural information on large protein complexes and for systems that exhibit multiple conformational or compositional states is gaining increasing importance. The cryo-EM approach, although limited in resolution, could also provide a link by being suitable for the study of systems in which protein is more limiting. The richness of structural biology can best be harnessed by combining various methods where practical and possible. The path forward thus appears to lie in the use of a combination of these techniques for structure elucidation, rather than heavy reliance on any single technique.
References:
- Stock D, Perisic O, Lowe J. Robotic Nanolitre Protein Crystallisation at the MRC Laboratory of Molecular Biology. Prog Biophys Mol Biol, 2005, 88(3): 311–327.
- John C. Chatham, Stephen J. Blackband, Nuclear Magnetic Resonance Spectroscopy and Imaging in Animal Research, ILAR Journal, Volume 42, Issue 3, 2001, Pages 189–208.
- Bai XC, McMullan G, Scheres SH. How cryo-EM is revolutionizing structural biology. Trends Biochem Sci. 2015 Jan;40(1):49-57.
- Egli M. Diffraction techniques in structural biology. Curr Protoc Nucleic Acid Chem. 2010;Chapter 7:Unit-7.13.
- Vénien-Bryan C, Li Z, Vuillard L, Boutin JA. Cryo-electron microscopy and X-ray crystallography: complementary approaches to structural biology and drug discovery. Acta Crystallogr F Struct Biol Commun. 2017 Apr 1;73(Pt 4):174-183.
- Muench SP, Antonyuk SV, Hasnain SS. The expanding toolkit for structural biology: synchrotrons, X-ray lasers and cryoEM. IUCrJ. 2019 Mar 1;6(Pt 2):167-177.