Analyzing Purification for Optimal Bioseparation
Analyzing Purification for Optimal Bioseparation
Methods comparison; size-exclusion, ion-exchange and affinity chromatography
Introduction
Several biological products such as biochemicals, biopharmaceuticals, foods, nutraceuticals, and agrochemicals require extensive purification and analysis before they can be used for their respective applications. The efficient removal of interfering matrix components followed by fast and accurate quantitation of biomolecules has become increasingly important following any bioprocess. The practice of purifying biological analytes on a large-scale, using fundamental aspects of engineering and scientific principles is termed bioseparation. Separating complex mixtures into purified fractions containing specific molecules of interest is the end goal of bioseparation. It is a broader term than the slightly dated downstream processing which specifically referred to the separation and purification segment of a bioprocess which followed some form of biological reaction e.g. purification of an antibiotic following microbial fermentation. The purification of biological products from their respective starting material e.g. cell culture media is technically difficult and expensive. This could frequently be the critical limiting factor in the commercialization of a biological product.
Biomolecules such as carbohydrates, proteins and nucleic acids cannot withstand the rigors of traditional chemical separation techniques and require the use of special methods. For instance, organic solvents, which are widely used in chemical separations, have relatively limited usage in bioseparations on account of their tendency to promote degradation of many biological products. Bioseparation techniques have to be “gentle” in terms of avoiding extremes of physicochemical conditions such as pH and ionic strengths, hydrodynamic conditions such as high shear rates, and exposure to gas-liquid interfaces. Some biologically derived substances such as antibiotics and other low molecular weight compounds such as vitamins and amino acids are purified using conventional separation techniques such as liquid-liquid extraction, packed bed adsorption, evaporation and drying with practically no modifications being necessary. However, substantially modified separation techniques are required for purifying more complex molecules such as proteins, lipids, carbohydrates and nucleic acids. Often, totally new types of separation techniques have to be devised.
Biological products are separated and purified based on one or more of the following properties:
- Size: e.g. filtration, membrane separation, centrifugation
- Density: e.g. centrifugation, sedimentation, floatation
- Diffusivity: e.g. membrane separation
- Shape: e.g. centrifugation, filtration, sedimentation
- Polarity: e.g. extraction, chromatography, adsorption
- Solubility: e.g. extraction, precipitation, crystallization
- Electrostatic charge: e.g. adsorption, membrane separation, electrophoresis
- Volatility: e.g. distillation, membrane distillation, pervaporation
Bioseparation is frequently based on multi-technique separation. Typically, low-resolution techniques (e.g. precipitation, filtration, centrifugation, and crystallization) are used first, for recovery and isolation, as they significantly reduce the volume of material being processed. This is followed by high-resolution techniques (e.g. affinity separations, chromatography, and electrophoresis), for purification and polishing. Three key analytical and purification methods are: electrophoresis, ultracentrifugation, and chromatography. Each one relies on certain physicochemical properties of different biomolecules.
Electrophoresis:
Many important biological molecules such as proteins, deoxyribonucleic acid (DNA), and ribonucleic acid (RNA) exist in solution as cations or anions. Under the influence of an electric field, these molecules migrate at a rate that depends on their net charge, size and shape, the field strength, and the nature of the medium in which the molecules are moving. Thus, separation is based on both the molecular sieve effect and on the electrophoretic mobility of the molecules. This method determines the size of biomolecules. It is used to separate proteins, and especially to separate DNA for identification, sequencing, or further manipulation.
Ultracentrifugation:
Cells, organelles, or macromolecules in solution exposed to a centrifugal force will separate because they differ in mass, shape, or a combination of those factors. An ultracentrifuge generates centrifugal forces of 600,000 g and more. It is an indispensable tool for the isolation of proteins, DNA, and subcellular particles.
Chromatography:
Chromatography encompasses a diverse and important group of methods that allow the separation, identification, and determination of closely related components of complex mixtures. Chromatographic separation entails separation of sample components based on differential affinity for a mobile versus a stationary phase. The mobile phase is a liquid or a gas that flows over or through the stationary phase, which consists of spherical particles packed into a column. When a mixture of proteins is introduced into the mobile phase and allowed to migrate through the column, separation occurs because proteins that have a greater attraction for the solid phase migrate more slowly than do proteins that are more attracted to the mobile phase. Several different types of interactions between the stationary phase and the substances being separated are possible and hence various parameters can be used to categorize chromatographic methods (Table 1).
Table 1: c:
| Parameters | Principle of Separation | Mobile Phase Used | Geometry of Technique | Scale of Operation | Mode of Technique | Elution Method |
|---|---|---|---|---|---|---|
| T | Partition | GC | Planar-paper, TLC, HPTLC | Preparative | Normal phase | Gradient |
| Y | Adsorption | LC | Planar-paper, TLC, HPTLC | Preparative | Normal phase | Gradient |
| P | Size-exclusion | Super Critical Fluid C | Column-GC, HPLC, SFC, UPLC | Analytical | Reverse phase | Isocratic |
| E | Affinity | Super Critical Fluid C | Column-GC, HPLC, SFC, UPLC | Analytical | Reverse phase | Isocratic |
| S | Ion-exchange | Super Critical Fluid C | Column-GC, HPLC, SFC, UPLC | Analytical | Reverse phase | Isocratic |
Table 1: Classification of chromatographic techniques based on various parameters.
Paper chromatography
In paper chromatography the support material consists of a layer of cellulose highly saturated with water. In this method a thick filter paper comprises the support, and water drops settled in its pores make up the stationary “liquid phase.” Mobile phase consists of an appropriate fluid placed in a developing tank. Paper chromatography is a “liquid-liquid” chromatography (Fig 1). It is primarily used to separate colored substances.
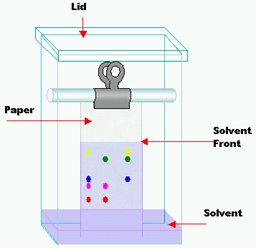
Fig 1: Paper chromatography
Gas Chromatography
In this method the stationary phase is a column which is placed in the device, and contains a liquid stationary phase which is adsorbed onto the surface of an inert solid. Gas chromatography is a “gas-liquid” chromatography. Its carrier phase consists of gases such as He or N2. An inert gas, which forms the mobile phase, is passed through a column under high pressure. The sample to be analyzed is vaporized, and enters into this gaseous mobile phase. The components contained in the sample are dispersed between the mobile phase, and the stationary phase on the solid support. Gas chromatography is a simple, multifaceted, highly sensitive, and rapidly applied technique for the extremely excellent separation of very minute molecules. It is used in the separation of very small amounts of analytes.
Thin Layer Chromatography
Thin-layer chromatography is a “solid-liquid adsorption” chromatography. In this method the stationary phase is a solid adsorbent substance coated on glass plates. As the adsorbent material all solid substances used in column chromatography (alumina, silica gel, cellulose) can be utilized. In this method, the mobile phase travels upward through the stationary phase. The solvent travels up the thin plate soaked with the solvent by means of capillary action. During this process, it also drives the mixture, priorly dropped on the lower parts of the plate with a pipette, upwards with different flow rates. Thus the separation of analytes is achieved. This upward travelling rate depends on the polarity of the material, the solid phase, and that of the solvent.
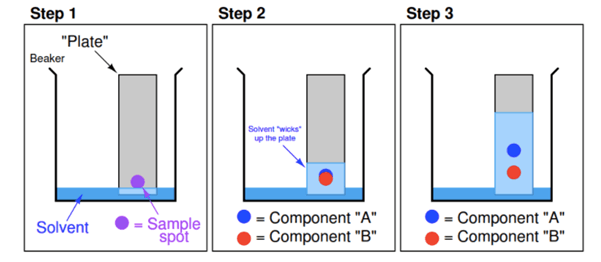
Fig. 2. Thin layer chromatography.
Ion-exchange Chromatography
Ion-exchange chromatography is based on the electrostatic interactions between charged protein groups, and solid support material (matrix). The matrix has an ion load opposite to that of the protein to be separated, and the affinity of the protein to the column is achieved with ionic ties. Proteins are separated from the column either by changing pH, concentration of ion salts or ionic strength of the buffer solution. Positively charged ion-exchange matrices are called anion-exchange matrices and they adsorb negatively charged proteins. While matrices bound with negatively charged groups are known as cation-exchange matrices, and they adsorb positively charged proteins.
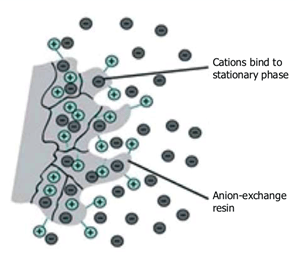
Fig. 3. Ion exchange chromatography.
Gel Filtration Chromatography
Gel filtration (also called size-exclusion chromatography or SEC) uses a porous resin material to separate molecules based on size (i.e., physical exclusion). The sample is applied to the top of a column consisting of porous beads made of an insoluble but highly hydrated polymer such as dextran or agarose (which are carbohydrates) or polyacrylamide. Sephadex, Sepharose, and Bio-gel are commonly used commercial preparations of these beads, which are typically 100 μm (0.1 mm) in diameter. Small molecules can enter these beads, but large ones cannot. The result is that small molecules are distributed in the aqueous solution both inside the beads and between them, whereas large molecules are located only in the solution between the beads. Large molecules flow more rapidly through this column and emerge first because a smaller volume is accessible to them. Molecules that are of a size to occasionally enter a bead will flow from the column at an intermediate position, and small molecules, which take a longer, tortuous path, will exit last.

Fig. 4. Size-exclusion chromatography.
Hydrophobic Interaction Chromatography (HIC)
In hydrophobic interaction chromatography, hydrophobic residues on the protein’s surface interact with hydrophobic ligands attached to a hydrophilic chromatographic resin. The type of hydrophobic ligand and its concentration on the resin are important parameters that control its separation properties. The type of hydrophobic ligand, for example the varying links of alkyl chains, in its concentration on the resin are important parameters of the resin that control its hydrophobicity and separation properties. In HIC, the interaction of the protein with the resin is modulated by adding or removing anti-chaotropic salts, such as ammonium sulfate, to the buffer. High salt concentrations promote adsorption to the hydrophobic groups in the column. Therefore, to elute adsorbed proteins for the column, the salt concentration is decreased. As the salt concentration is decreased, proteins of increasing hydrophobicity will desorb and elute from the column. HIC is often most useful as a polishing step to remove higher molecular weight impurities that may not be removed by other methods.
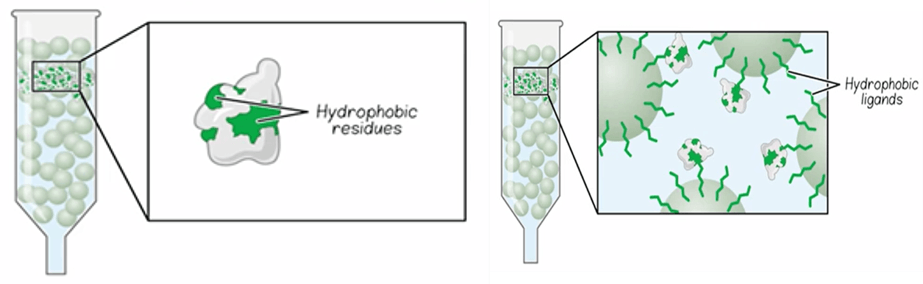
Fig. 5. Hydrophobic interaction chromatography.
Affinity Chromatography
Affinity chromatography is one of the oldest forms of liquid chromatography. The first use may be considered as the isolation of α-amylase by using an insoluble substrate, starch, in 1910 just three years after the discovery of chromatography. This chromatographic technique has become increasingly important in work with pharmaceutical agents, biochemical and biological samples, clinical chemistry and environmental science.
It is one of the most diverse and powerful chromatographic method for the purification of a specific molecule or a group of molecules from a complex mixture by the selective non-covalent interaction of solute with a ligand (which is immobilized). The wide applicability of this method is based on the fact that any given biomolecule that one wishes to purify usually has an inherent recognition site through which it can be bound by a natural or artificial molecule. Thus, we can say that affinity chromatography is principally based on the molecular recognition of a target molecule by a molecule bound to a column.
Application of affinity chromatography has significant advantages. The important one is that affinity chromatography involves many types of interactions between ligand and target such as steric effects, hydrogen bonding, ionic interactions, van der Waals forces, dipole-dipole interactions and even covalent bonds while other chromatographic techniques involve just one or a few of them. The combination of these multiple interactions leads to separation with high selectivity and retention in affinity chromatography. In addition to its solute purification potential, affinity chromatography also possesses considerable potential for investigating the functional roles of the reactants purified.
Affinity purification involves three main steps (Fig. 6):
- Incubation of a crude sample with the affinity support to allow the target molecule in the sample to bind to the immobilized ligand.
- Washing away non-bound sample components from the support.
- Elution (dissociation and recovery) of the target molecule from the immobilized ligand by altering the buffer conditions so that the binding interaction no longer occurs.

Fig. 6. Affinity chromatography.
Affinity Ligands
In order to distinguish the techniques according to the origin of the ligand, affinity chromatography with biological ligands may be termed as “bioaffinity chromatography” or “biospesific adsorption”.
Some typical biological interactions frequently used in affinity chromatography are:
- Enzyme ↔ substrate analogue, inhibitor, cofactor.
- Antibody ↔ antigen, virus, cell.
- Lectin ↔ polysaccharide, glycoprotein, cell surface receptor, cell.
- Nucleic acid ↔ complementary base sequence, histones, nucleic acid polymerase, nucleic acid binding protein.
- Hormone, vitamin ↔ receptor, carrier protein.
- Glutathione ↔ glutathione-S-transferase or GST fusion proteins.
Selectivity of the ligand, recovery process, throughput, reproducibility, stability and economic criteria are some of the factors that influence the success of the affinity chromatography process. Choice of the support material, ligand, activation method and conditions for adsorption and desorption are important considerations prior to setting up a typical affinity chromatography experiment.
- Support material: The support within the column must contain an affinity ligand that is capable of forming a suitably strong complex with the solute of interest. Also, the support material must be biological and chemically inert. There are many commercially available support materials for affinity chromatography which can be divided into three groups: natural (agarose, dextrose, cellulose); synthetic (acrylamide, polystyrene, polymethylacrylate) and inorganic (silica, glass) materials.
- Ligand: The selected ligand must be capable of selectively and reversibly binding to the substance to be isolated and have some groups which are available for modifications in order to be attached to the support material. It is very important to ensure that the modifications do not reduce the specific binding affinity of the ligands. There are general ligands such as dyes, amino acids, Protein A and G, lectin, coenzyme, metal chelates as well as specific ligands such as enzymes and substrates, antibodies and antigens.
Affinity ligands are classified as synthetic and biological. Biological ligands are obtained from natural sources such as RNA and DNA fragments, nucleotides, coenzymes, vitamins, lectins, antibodies, binding or receptor proteins, or in vitro from biological and genetic packages, employing display techniques including oligonucleotides, peptides, protein domains and proteins.
Synthetic ligands are generated using three methods:
- The rational method features the functional approach and structural template approach.
- The combinatorial method relies on the selection of ligands from a library of synthetic ligands synthesized randomly.
- The combined method employs both methods and the ligand is selected from an intentionally prepared library based on a rationally designed ligand.
Many parameters have to be taken into account in order to select and design an appropriate ligand.
- Determination of the binding site or possible biological interactions to use as a template for the modelling,
- Initial design of the ligand using this template,
- Preparation of a ligand library and chromatographic evaluation,
- Selection of the ligand of interest,
- Optimization and chromatographic evaluation of the adsorbent following the immobilization.
Table 2 exhibits the advantages and disadvantages of biological and synthetic ligands:
| Synthetic ligands | Biological ligands | |
|---|---|---|
| Capacity | High | Low medium |
| Cost | Low to medium | Medium to high |
| Selectivity | Medium to high | Very high |
| Stability | High | Low to medium |
| Toxicity | Medium | Low |
Table 2: Advantages and disadvantages of synthetic and biological ligands.
Selectivity and affinity are the main advantages of biological ligands. Such ligands can be generated by in vitro evolution approaches and selecting from large combinatorial ligand libraries based on biological/genetic packages. Protein ligands display special advantages for example; higher affinities, higher proteolytic stability, preservation of their biological activity when produced by fusion to a different protein or domain. However these ligands can be expensive and unstable to the sterilization and cleaning conditions used in manufacturing processes of biologics because of their biological origin, chemical nature and production methods.
The wide application potential of affinity chromatography led to the development of derived techniques some of which are listed below:
- High performance affinity chromatography
- Dye-ligand affinity chromatography
- Lectin affinity chromatography
- Immunoaffinity chromatography
- Metal-chelate affinity chromatography
- Covalent affinity chromatography
Immunoaffinity chromatography (IAC):
Immunoaffinity chromatography (IAC) is a very popular technique that enables the production of ligands in case the ligand required is not available. In this technique, the stationary phase comprises of an antibody or antibody-related agent. It is possible to isolate a variety of substances using this technique due to the high specificity of antibodies. It is reported that IAC may be used for natural food contaminants such as aflatoxins, fumonisins and ochratoxins. IAC is probably the most highly specific of all forms of bioaffinity chromatography and both large and small analytes can be purified using this technique.
Lectin affinity chromatography:
Lectin affinity chromatography Lectins are non-immune proteins produced by plants, vertebrates and invertebrates. Especially, various plant seeds synthesize high levels of lectins. Certain types of carbohydrate residues may be separated via this method as all lectins have the ability to recognize and bind these types of compounds. Mostly used lectins for affinity columns are concanavalin A, soybean lectin and wheat germ agglutinin. Concanavalin A is specific for α-D-mannose and α-D-glucose residues while wheat germ agglutinin binds to D-N-acetyl-glucosamine.
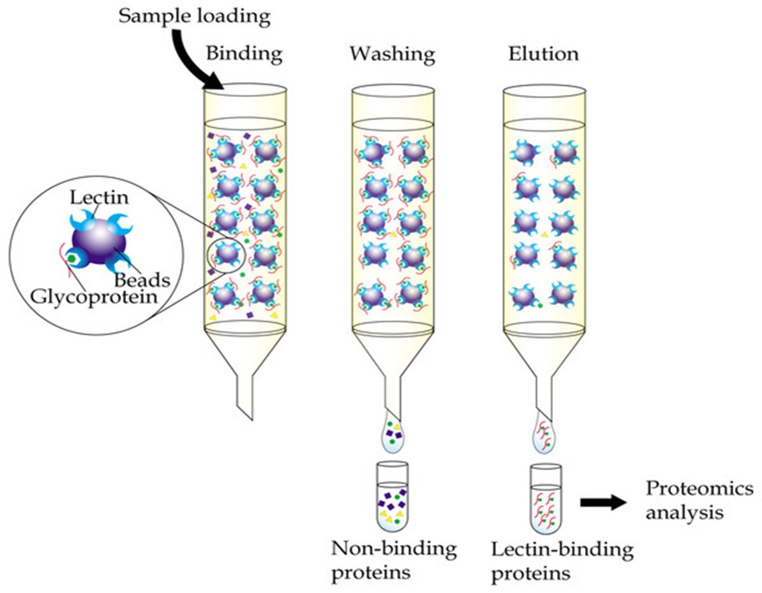
Fig.7. Lectin affinity chromatography.
Metal chelate affinity chromatography (Immobilized metal ion affinity chromatography or IMAC):
Metal chelate affinity chromatography (Immobilized metal ion affinity chromatography or IMAC) is based on the ability of certain amino acids acting as electron donors on the surface of proteins (histidine, tryptophan, tyrosine, or phenylalanine) to bind reversibly to transition metal ions that have been immobilized by a chelating group covalently bound to a solid support. Of these amino acids, histidine is quantitatively the most important in mediating the binding of most proteins to immobilized metal ions. Histidine binds selectively to immobilized metal ions even in the presence of excess free metal ions in solution. Copper and nickel ions have the greatest affinity for histidine. The affinity of histidine residues for immobilized Ni2+ ions allows selective purification of proteins containing a high proportion of histidine residues on the surface. Nitrilotriacetic acid (NTA), has provided a convenient and inexpensive support for purification of proteins containing histidine residues.
Thus, chromatography plays a crucial role in the drug discovery and development process. In fact, the process of drug discovery and development would be incomplete without the use of appropriate chromatographic techniques for purification and analysis. The choice of a suitable chromatographic method coupled with an adequate detection technique, depending on the nature of the analyte, can greatly reduce the time and cost involved in all steps from the drug discovery to the manufacturing stage.
References:
- Luxminarayan, L., Sharma, N., Viswas, A., Viswas, A., & Khinchi, D. (2017). A review on chromatography techniques. Asian Journal of Pharmaceutical Research and Development, 5(2), 1-08.
- Karlsson E, Ryden L, Brewer J. Protein Purification. Principles, High Resolution Methods, and Applications, 2nd Edition, Ion exchange chromatography. Wiley-VCH, New York;1998.
- Coskun O. Separation techniques: Chromatography. North Clin Istanb. 2016;3(2):156-160.
- Porath J. Immobilized metal ion affinity chromatography. Protein Expr Purif. 1992;3:263–81.
- Wilchek M, Chaiken I. An overview of affinity chromatography in affinity chromatography–Methods and protocols. Humana Press. 2000:1–6.
- Determann H. Gel chromatography gel filtration, gel permeation, molecular sieves:a laboratory handbook. Chapter 2. Materials and Methods. 2012
- Petty KJ. Metal-chelate affinity chromatography. Curr Protoc Mol Biol. 2001 May;Chapter 10:Unit 10.11B.